I'm an NSF postdoctoral fellow in the Martin lab at UC Berkeley, studying the genomic basis of fitness differences underlying the adaptive radiation of trophic specialist pupfish on San Salvador Island.
Assessing the role of hybridization in macroevolutionary diversification
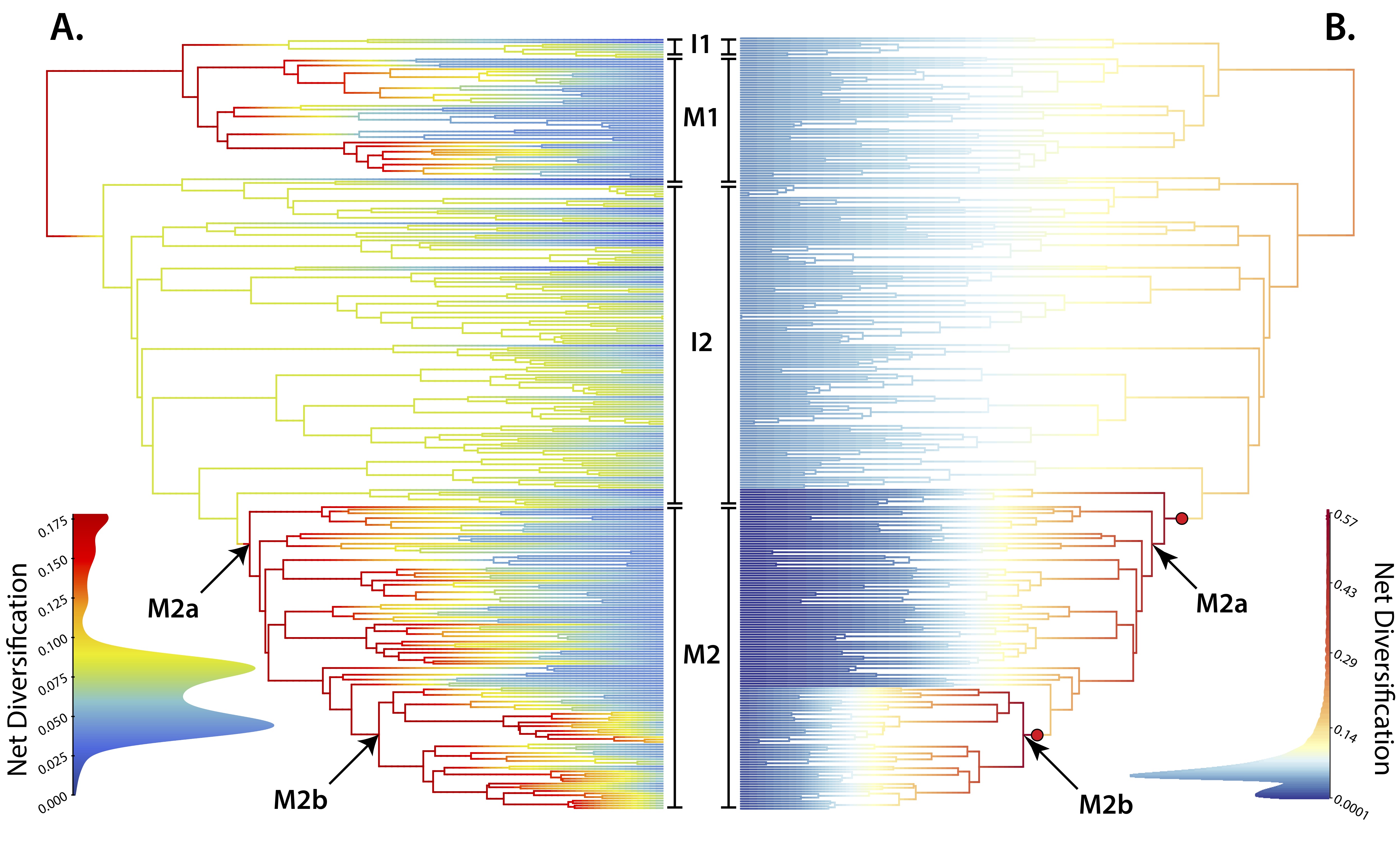
Whether hybridization generates or erodes species diversity has long been debated, but to date most studies have been conducted at small taxonomic scales. Salamanders (order Caudata) represent a taxonomic order in which hybridization plays a prevalent ecological and evolutionary role. In this study, we employ a recently developed model of trait-dependent diversification, HiSSE, to test the hypothesis that speciation and extinction rates will differ between hybridizing and non-hybridizing taxa. We find strong evidence supporting this hypothesis, showing that hybridizing salamander lineages have significantly greater net-diversification rates than non-hybridizing lineages. This pattern is driven by concurrently increased speciation rates and decreased extinction rates in hybridizing lineages. Our results support the hypothesis that hybridization can act as a generative force in macroevolutionary diversification. A video of my recent presentation at Evolution 2017 on this research is posted below.
This work is in collaboration with Andrew Storfer (PI), Luke Harmon, Mark Margres, Brendan Epstein, and Jon Eastman.
Adaptive radiation has long captured the interest of evolutionary biologists. Defined as the accumulation of ecologically differentiated, coexisting species within a rapidly diversifying lineage, adaptive radiations have long been treated as model systems to study the intersection of ecology and evolution. Despite decades of study however, there exists little consensus regarding what governs the tempo and mode of adaptive radiation. Often occurring following colonization of a new, geographically isolated and ecologically distinct landscape, adaptive radiations have most frequently been described and studied in island-like systems. The consequence of this tendency has led to a paradigm in which adaptive radiations are almost synonymized with islands and lakes. In this study, we show that colonization of upper Central America by Anolis lizards led to their rapid radiation on the mainland. Importantly, timing of colonization plays a crucial role in the eventual outcome of diversification; Central America and the Nearctic was at the time isolated from South America and harbored no Anoline diversity, thus enabling this radiation. We find that although colonization can lead to dramatic shifts in diversification dynamics, patterns of morphological rate variation are decoupled from lineage diversification rates, yet predict morphological disparity. Further, we present evidence of widespread convergence in mainland anoles, exceeding that observed within island lineages. Our study emphasizes the need to focus research efforts on the study of historically understudied mainland diversity. Although radiations on continental landmasses are typically less readily circumscribed, our study demonstrates that these radiation may harbor equally, if not more exceptional diversity than their island counterparts.
This work is in collaboration with Luke Harmon, Luke Mahler, Anthony Herrel, and Jonathan Losos.
Comparative transcriptomics of hybridization
The Southern Appalachians contain the greatest diversity of salamanders in the world. Of the salamanders native to this region, the Plethodontid (terrestrial lungless) salamanders are the most diverse, and many of these have been observed to hybridize in nature. In some cases, hybrizing species pairs hybridize in some locations but not in others. This appreciable frequency of hybridization thus lends to the question of what the consequences of hybridization are for these species. One means by which we may address this is through the use of transcriptomics, thus characterizing patterns of gene expression across parental taxa, relative to hybrids. We have assembled and are currently annotating the (tail) transcriptomes of three species of eastern plethodontid salamanders (Plethodon jordani, Plethodon metcalfi, Plethodon teyahalee) that hybridize frequently in nature. In an upcoming series of projects, we aim to leverage these transcriptomes and those of their hybrids to identify genomic regions associated with differential expression, both among parental taxa and aon hybrids. By relating these differentially expressed genes back to their functional annotations, we thus may gain insight into 1) what genes lead to interspecific differences, and 2) aspects of the transcriptome most frequently impacted by hybridization among taxa.
This work is in collaboration with Andrew Storfer (PI), Joanna Kelley and Mark J. Margres.
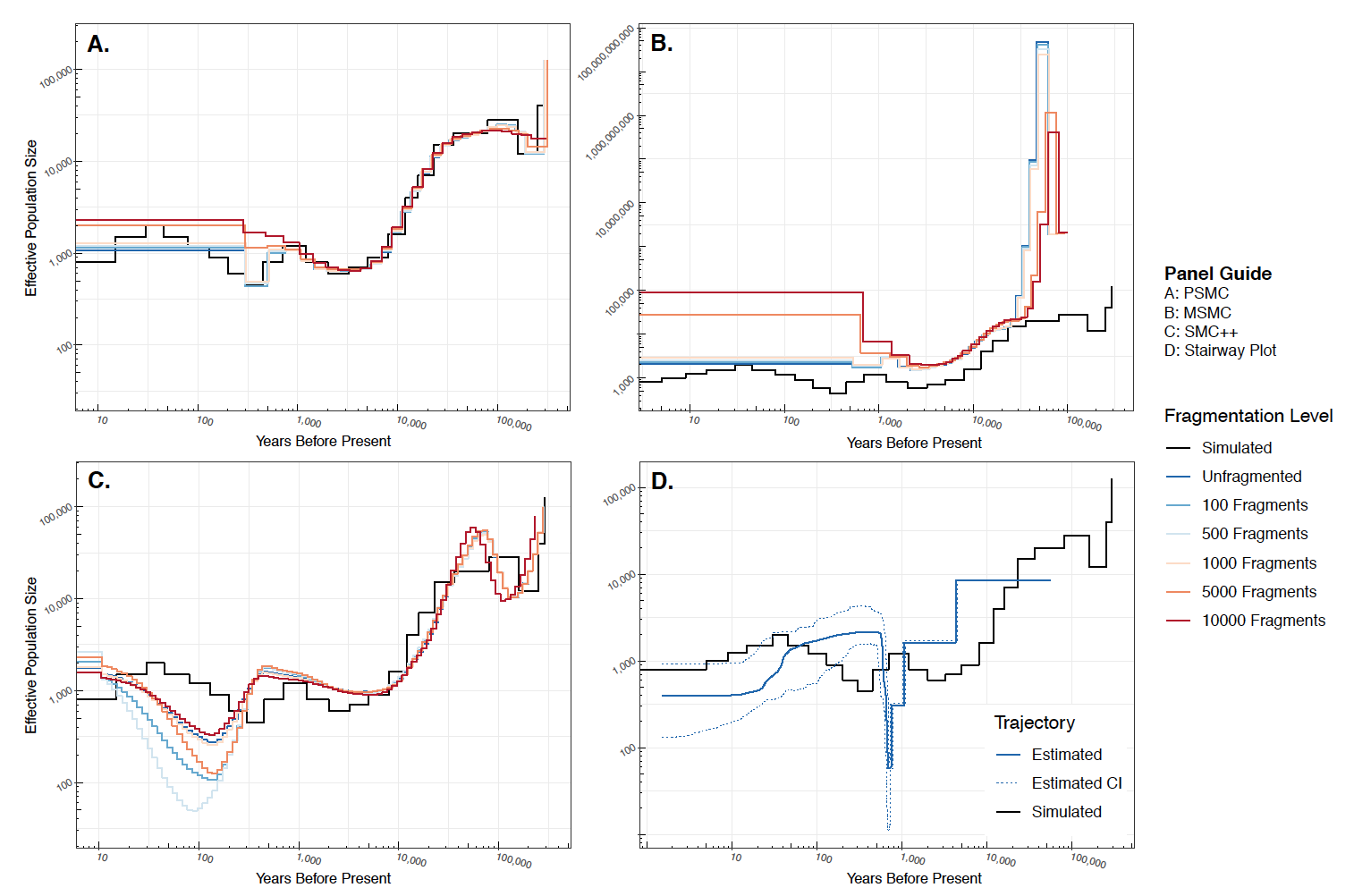
Comparison of contemporary demographic reconstruction methods
Reconstructing species' demographic histories is a central focus of molecular ecology and evolution. Recently, an expanding suite of methods leveraging either the sequentially Markovian coalescent (SMC) or the site-frequency spectrum (SFS) have been developed to reconstruct population size histories from genomic sequence data. However, few studies have investigated the robustness of these methods to genome assemblies of varying quality. In this study, we presented an improved genome assembly for the Tasmanian devil using the Chicago library method. Compared to the original reference genome, our new assembly reduced the number of scaffolds (from 35,975 to 10,010) and increased the scaffold N90 (from 0.101 to 2.164 Mb). Second, we assessed the performance of four contemporary genomic methods for inferring population size history (PSMC, MSMC, SMC++, Stairway Plot), using the two devil genome assemblies as well as simulated, artificially fragmented genomes that approximate the hypothesized demographic history of Tasmanian devils. We demonstrated that each method is robust to assembly quality, producing similar estimates of Ne up to around 5,000 fragments. Overall, methods reliant on the SMC are most reliable between ~300 generations before present (gbp) and 100 kgbp, whereas methods exclusively reliant on the SFS are most reliable between the present and 30 gbp. Our results suggest that whole-genome methods for reconstructing species' effective population size histories: 1) can be applied to non-model organisms without highly contiguous reference genomes; 2) are capable of detecting independently documented effects of historical geological events; and, 3) should be used in concert.
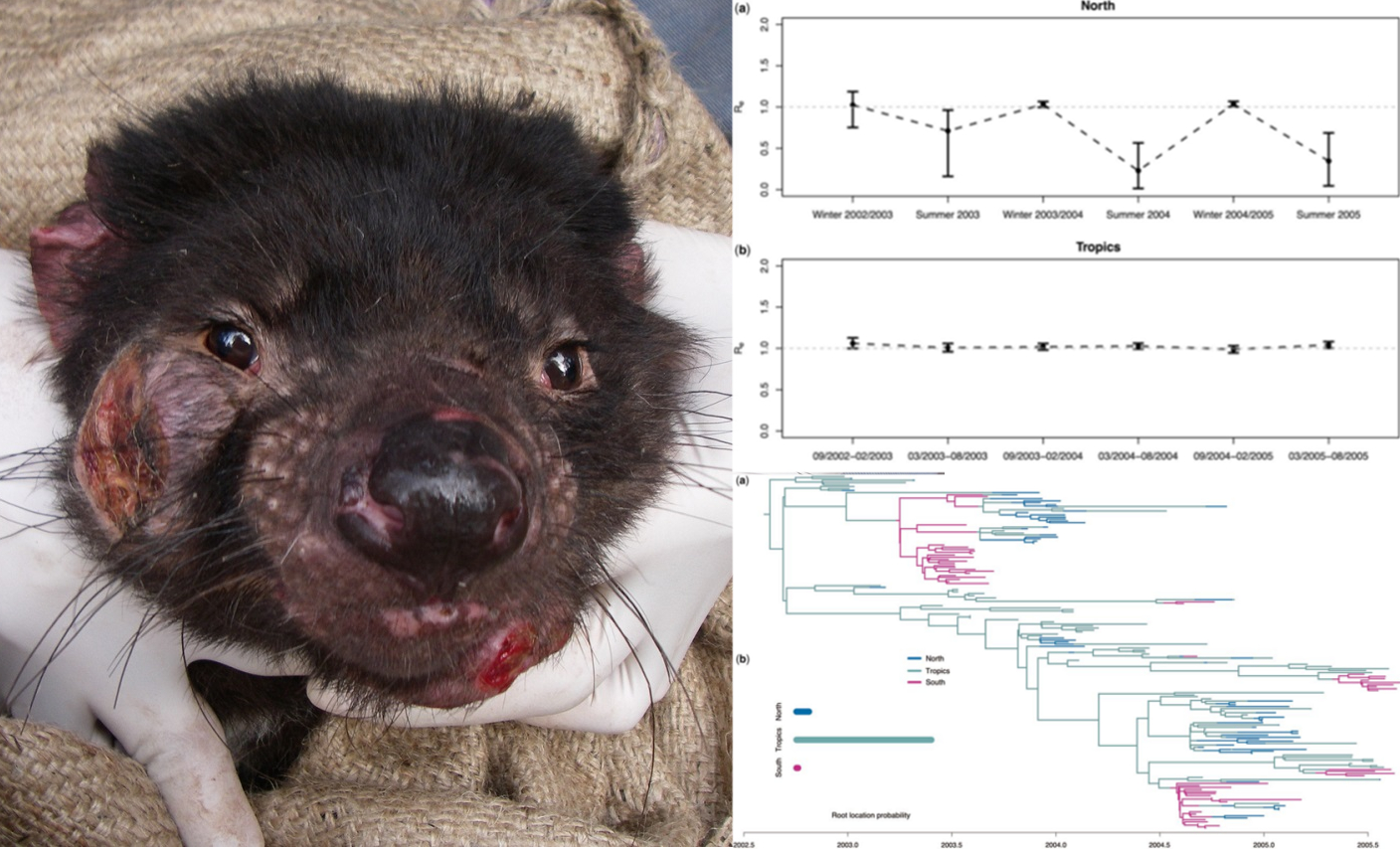
Phylodynamics of a transmissible cancer
Of paramount importance to the conservation of species diversity is an understanding of emerging infectious diseases (EIDs). Frequently driven by novel interactions among previously isolated species, EIDs have been implicated in recent declines or even extinctions in the cases of Dutch elm disease, chytrid fungus in amphibians, and avian malaria an Hawaiian birds. Given the increased prevalence of habitat alteration and severity of global climate change, geographic ranges of many species, including vectors of EIDs are expected to change dramatically. Such range shifts have the potential to facilitate both the spread and establishment of EIDs, as was observed recently with the emergence of Zika virus in Brazil of early 2015. In this study, we aim to characterize the transmission dynamics among lineages of one such EID, the Tasmanian Devil Facial Tumor Disease (DFTD), using a phylodynamic approach. Leveraging low-coverage whole-genome sequencing of 50 serially-sampled tumors collected from Tasmanian Devils Sarcophilus harrisii, we will be able to identify tumor strains exhibiting greater virulance. With this information, we then may begin to identify those components of DFTD evolution that led to these increased lineage fitnesses.
This work is in collaboration with Andrew Storfer (PI), Hamish McCallum (Co-PI), Menna Jones (Co-PI), Paul Hohenlohe, and Mark Margres.